![]()
![]()
![]()
![]()
Here you will find our mature and stable software tools with user support.
For the identification of peptides in a sample by tandem mass spectra, as generated by data-dependent acquisition, protein sequence databases like the well known UniProtKB, provide the basis for most spectrum identification search engines. In addition, for targeted proteomics approaches like selected reaction monitoring and parallel reaction monitoring, knowledge of the peptide sequences, their masses, and whether they are unique for one protein in a database is essential. Because most bottom-up proteomics approaches use trypsin to cleave the proteins in a sample, the tryptic peptides contained in a protein database are of great interest. MaCPepDB (mass-centric peptide database) consists of the complete tryptic digest of the Swiss-Prot and TrEMBL parts of UniProtKB. This database is especially designed to query peptides by sequence or mass with additional filters, like mass tolerance or posttranslational modifications, and return the respective annotated peptides.

Publications
Downloads
Contact
Links
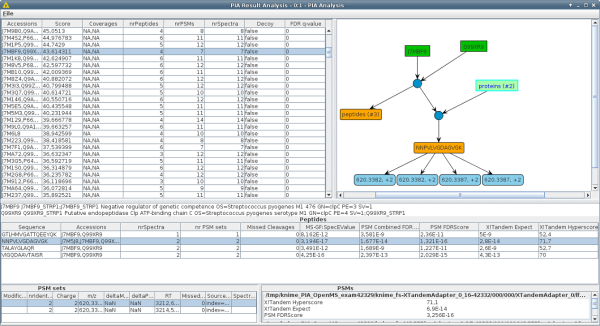
PIA is a toolbox for MS-based protein inference and identification analysis. With PIA, you can view the results of popular mass spectra identification search engines, combine them, and perform statistical analyses. The main focus of PIA is on the integrated inference algorithms, i.e. inferring proteins from a set of identified peptides. However, it also allows to inspect peptide spectrum matches (PSMs), calculate FDR values across different search engine results and visualize the correspondence between PSMs, peptides and proteins.
PIA in a Nutshell
Most search engines for protein identification in MS/MS experiments return protein lists, even though the actual search returns a set of peptide spectrum matches (PSMs). The step from PSMs to proteins is called "protein inference." When a set of identified PSMs supports detection of more than one protein in the searched database ("protein ambiguity"), only one representative protein is usually reported. These proteins may differ depending on the search engine and settings used, so protein lists from different search engines cannot usually be compared. PSMs from complementary search engines are often combined to increase the number of reported proteins or to verify the evidence of a peptide improved by detection with different algorithms.
We have developed PIA, an algorithm suite written in Java that contains fully parameterizable KNIME nodes that combine PSMs from different experiments and/or search engines to produce consistent and thus comparable results. Thereby, different parameters e.g. regarding filtering can be flexibly adjusted. PIA can be invoked from the command line (even in Docker containers) or in the KNIME workflow environment, allowing seamless integration with OpenMS workflows.
Publications
Downloads
Contact
Links
The BIONDA Biomarker Database provides structured information on all biomarker candidates published in PubMed articles. There is no limitation to any type of disease. To this end, PubMed article abstracts and reputable databases such as UniProt and Human Disease Ontology are used as sources for BIONDA's database entries. These are automatically collected and regularly updated using text mining methods. BIONDA is freely available via a user-friendly web interface. As a specific feature, BIONDA's database entries are evaluated by a scoring approach that estimates biomarker reliability.
Publication
Contact
Links

CalibraCurve is an R package designed to generate calibration curves for targeted mass spectrometry-based quantitative data. It is applicable to various omics disciplines, including proteomics, lipidomics, and metabolomics. The package also offers functionalities for data and calibration curve visualization and concentration prediction from new datasets based on the established curves.
Publication
Download
Contact
Copyright © MPC 2026
Last update: Jan 28, 2026